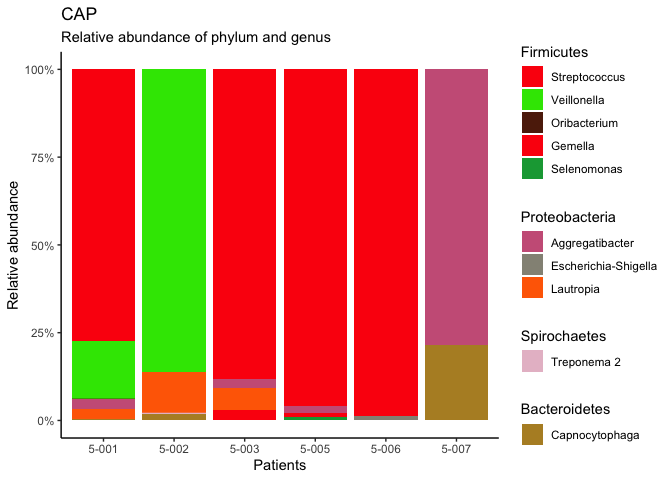
Im want make better the legend()
I am looking for the legend to be grouped according to a higher classification (phylum) but that at the same time the genus (Genus) is shown.
I want to have something like that: Im try to make something like this
{kind=link}
Im run this code:
ggplot(d4)
geom_bar(aes(x=Pacientes, y=`Relative abundance`,fill=Genus), position="fill", stat="identity") scale_x_discrete("Patients")
scale_y_continuous("Relative abundance",labels=scales::percent)
labs(title = "CAP",subtitle = "Relative abundance of phylum and genus")
theme_classic()
scale_fill_manual(values=c ("#FC000D", "#30E500", "#E10072", "#730183", "#B58E2C","#10A542","#6C1429",
"#00B9B9", "#E36582","orange3","#800009",
"#5E230B","#CC6187","#949285","#FF6A00",
"#FF9D69","#B08A04","#005A3F","#120A5F","#E7BECD"))
{kind=link}
Example data
d4<-structure(list(Pacientes = c("5-006", "5-005", "5-005", "5-001",
"5-003", "5-002", "5-001", "5-001", "5-005", "5-001", "5-003",
"5-003", "5-007", "5-006", "5-003", "5-001", "5-002", "5-003",
"5-002", "5-002", "5-001", "5-002", "5-003", "5-005", "5-002",
"5-001", "5-006", "5-005", "5-007", "5-005"), Filum = c("Firmicutes",
"Firmicutes", "Firmicutes", "Firmicutes", "Firmicutes", "Firmicutes",
"Firmicutes", "Firmicutes", "Proteobacteria", "Proteobacteria",
"Proteobacteria", "Proteobacteria", "Proteobacteria", "Proteobacteria",
"Proteobacteria", "Proteobacteria", "Proteobacteria", "Proteobacteria",
"Spirochaetes", "Spirochaetes", "Spirochaetes", "Spirochaetes",
"Firmicutes", "Firmicutes", "Bacteroidetes", "Bacteroidetes",
"Bacteroidetes", "Bacteroidetes", "Bacteroidetes", "Firmicutes"
), Genus = structure(c(1L, 1L, 1L, 1L, 1L, 2L, 2L, 12L, 13L,
13L, 13L, 13L, 13L, 14L, 15L, 15L, 15L, 15L, 20L, 20L, 20L, 20L,
21L, 21L, 25L, 25L, 25L, 25L, 25L, 26L), .Label = c("Streptococcus",
"Veillonella", "Haemophilus", "Actinobacillus", "Serratia", "Fusobacterium",
"Neisseria", "Moraxella", "Abiotrophia", "Granulicatella", "Actinomyces",
"Oribacterium", "Aggregatibacter", "Escherichia-Shigella", "Lautropia",
"Geobacillus", "Leptotrichia", "Johnsonella", "Campylobacter",
"Treponema 2", "Gemella", "Megasphaera", "Atopobium", "Bifidobacterium",
"Capnocytophaga", "Selenomonas", "Mycoplasma", "Porphyromonas",
"Alloprevotella", "Lachnoanaerobaculum", "Eikenella", "[Eubacterium] brachy group",
"Stomatobaculum", "Atopostipes", "Selenomonas 3", "Kingella",
"Dialister", "F0058", "Parvimonas", "No identificado", "Solobacterium",
"Otros finales", "Olsenella", "Filifactor", "Rodentibacter",
"Alloscardovia", "Otros", "[Eubacterium] yurii group", "Anaeroglobus",
"Staphylococcus", "Ruminococcaceae UCG-014", "Lactobacillus",
"Rothia", "Selenomonas 4", "Scardovia", "Fluviicola", "Cardiobacterium",
"Bilophila", "Simonsiella", "[Eubacterium] nodatum group", "Catonella",
"Peptoniphilus", "uncultured", "Shuttleworthia", "Butyrivibrio 2",
"Peptostreptococcus", "Mogibacterium", "Bergeyella", "Peptococcus",
"Faucicola", "Blautia", "Rikenellaceae RC9 gut group", "Eggerthia",
"Desulfobulbus", "Tannerella", "Lactococcus", "Prevotella", "Otro",
"Prevotella 7", "Erysipelotrichaceae UCG-006", "Defluviitaleaceae UCG-011",
"W5053", "Craurococcus", "Dolosigranulum", "Sneathia", "Anaerococcus",
"Pseudoramibacter", "Family XIII UCG-001", "Ruminococcus 2",
"Howardella", "Cryptobacterium", "Listeria", "Pantoea", "Akkermansia",
"Prevotella 6", "Macrococcus", "Paracoccus", "Comamonas", "TM7 phylum sp. oral clone FR058",
"Peptoanaerobacter", "Rubellimicrobium", "Fastidiosipila", "Brachymonas",
"Candidatus Tammella", "Slackia", "DNF00809", "Truepera", "Finegoldia",
"Erysipelotrichaceae UCG-004", "uncultured bacterium", "Bulleidia",
"Flexilinea", "Methylobacterium", "Propionivibrio", "Ochrobactrum"
), class = "factor"), `Relative abundance` = c(1.797989737427,
1.17051056033446, 0.967773967968912, 0.890190018788368, 0.875168325944855,
0.203636768715721, 0.190978038791412, 0.0011814814596022, 0.0411830680204194,
0.031674955321716, 0.018003527003462, 0.00559797167763897, 0.0032912697803204,
0.0189880948864639, 0.0386794525465004, 0.0337566131314913, 0.0279054668553661,
0.0242766309437308, 0.000168783065657456, 0.000112522043771638,
8.43915328287282e-05, 8.43915328287282e-05, 0.0290306872930825,
0.0263020277316203, 0.00458527328369423, 0.00393827153200732,
0.00351631386786368, 0.00210978832071821, 0.000900176350173101,
0.021744884958869)), row.names = c(NA, -30L), class = c("tbl_df",
"tbl", "data.frame"))
The grouping colum is Filum and the normal legend is Genus in the data.
Thanks
CodePudding user response:
One option to achieve your desired result would be via the ggnewscale package which allows for multiple scales and legends for the same aesthetic.
- Put your colors into a named vector which assign a color to each of your
Genus - Make a list which of
Filums which associatedGenuss. To this end I make use ofdplyr::distinctandsplit.
library(ggplot2)
library(ggnewscale)
library(dplyr)
cols <- c("#FC000D", "#30E500", "#E10072", "#730183",
"#B58E2C", "#10A542", "#6C1429",
"#00B9B9", "#E36582", "orange3", "#800009",
"#5E230B", "#CC6187", "#949285", "#FF6A00",
"#FF9D69", "#B08A04", "#005A3F", "#120A5F", "#E7BECD")
cols <- rep_len(cols, length.out = length(levels(d4$Genus)))
names(cols) <- levels(d4$Genus)
groups <- d4 %>%
distinct(Filum, Genus) %>%
# Add order of Filum and legends
mutate(order = as.numeric(forcats::fct_inorder(Filum))) %>%
split(.$Filum)
- For each
Filumadd ageom_colshowing the whole data and ascale_fill_manualwhere using thelimitsargument we only show theGenuss associated with thisFilum. Doing so all otherGenuss will be assigned toNAfor which we choose atransparentcolor.
To get the basic idea of what we are doing here is the code for just two groups:
ggplot(d4)
geom_col(aes(x = Pacientes, y=`Relative abundance`, fill = Genus), position = "fill")
scale_fill_manual(values = cols, limits = groups$Firmicutes$Genus, na.value = "transparent")
new_scale_fill()
geom_col(aes(x = Pacientes, y=`Relative abundance`, fill = Genus), position = "fill")
scale_fill_manual(values = cols, limits = groups$Bacteroidetes$Genus, na.value = "transparent")
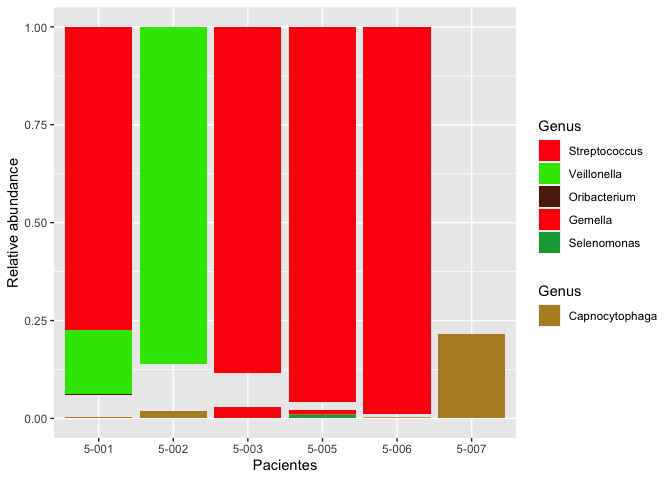
Instead of copying and pasting which becomes cumbersome if we have a lot of groups we could make use of lapply to loop over the groups and add the layers dynamically like so:
ggplot(d4)
lapply(groups, function(x) {
list(
geom_col(aes(x = Pacientes, y=`Relative abundance`, fill = Genus), position = "fill"),
scale_fill_manual(name = unique(x$Filum),
values = cols, limits = x$Genus, na.value = "transparent",
guide = guide_legend(order = unique(x$order))),
new_scale_fill()
)
})
scale_x_discrete("Patients")
scale_y_continuous("Relative abundance", labels = scales::percent)
labs(title = "CAP", subtitle = "Relative abundance of phylum and genus")
theme_classic()